
Naast morfologie zijn immunofenotypering, cytogenetische en moleculaire onderzoeken essentieel voor de juiste classificatie en indeling in risicogroepen van patiënten met AML.
Classificatie
WHO 2016 en 2022
WHO 2016
| WHO code |
Category | Subcategory and short description |
|---|---|---|
| *Rare cases show < 20% myeloblasts; these should be classified as AML | ||
| 9896 | AML with recurrent genetic abnormalities |
AML with t(8;21)(q22;q22); RUNX1-RUNX1T1* |
| 9871 | AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFß–MYH11* | |
| 9866 | Acute promyelocytic leukemia; APL with t(15;17)(q22;q12); PML–RARA and cytogenetic variants* | |
| 9897 | AML with t(9;11)(p22;q23); MLLT3-KMT2A | |
| 9865 | AML with t(6;9)(p23;q34); DEK-NUP214 | |
| 9869 | AML with inv(3)(q21q26.2) or t(3;3)(q21;q26.2); GATA2-MECOM | |
| 9911 | AML (megakaryoblastic) with t(1;22)(p13;q13); RBM15-MKL1 | |
| 9861 | AML with mutated NPM1 | |
| 9861 | AML with bi-allelic mutated CEBPA | |
| 9861 | Provisional entity: AML with BCR-ABL1 | |
| 9861 | Provisional entity: AML with mutated RUNX1 | |
| 9895 | AML with myelodysplasia related changes | Previous history of myelodysplastic syndrome or Myelodysplastic syndrome-related cytogenetic abnormality or Dysplasia present in > 50% of 2 or more cell lineages Absence of both Prior cytotoxic therapy for an unrelated disease Recurring cytogenetic abnormality as described in AML with recurrent genetic abnormalities |
| 9920 | Therapy-related myeloid neoplasms | Includes t-AML, t-MDS and t-MDS/MPN |
| 9861 | Acute myeloid leukemia, NOS | |
| 9872 | AML with minimal differentiation | <3% of blasts positive for Sudan Black B or MPO. Blasts usually express CD13 and/or CD117, with or without CD33 in absence of lymphoid markers cCD3, cCD22 and cCD79a |
| 9873 | AML without maturation | Blasts ≥90% of bone marrow non-erythroid cells (i.e. excluding also lymphocytes, plasmacells, macrophages and mast cells) ≥3% of blasts positive for Sudan Black B or MPO Blasts express MPO and one or more of myeloid-associated antigens such as CD13, CD33 or CD117 |
| 9874 | AML with maturation | ≥10% maturing cells of neutrophil lineage <20% bone marrow monocytes |
| 9867 | Acute myelomonocytic leukemia |
>20% neutrophils and precursors of marrow cells >20% monocytes and precursors of marrow cells |
| 9891 | Acute monoblastic and monocytic leukemia |
≥80% of the leukemic cells are monoblasts, promonocytes and monocytes |
| 9840 | Pure erythroid leukemia | >80% immature erythroid precursors with ≥30% pro-erythroblasts |
| 9910 | Acute megakaryoblastic leukemia |
>50% of the blasts are of megakaryocytic lineage Blasts express CD41 and/or CD61 |
| 9870 | Acute basophilic leukemia | Primary differentiation to basophils; mature basophils are usually sparse |
| 9931 | Acute panmyelosis with myelofibrosis |
Acute panmyeloid proliferation with accompanying fibrosis |
| 9930 | Myeloid sarcoma | Tumor mass of myeloblasts or immature myeloid cells occurring in an anatomical site other than the bone marrow |
| Myeloid proliferations related to Down syndrome (DS) | ||
| 9898 | Transient abnormal myelopoiesis (TAM) | |
| 9898 | Myeloid leukemia associated with Down syndrome | |
| 9727 | Blastic plasmacytoid dendritic cell neoplasm (BPDC) | |
| Blastic NK-cell lymphoma | ||
WHO 2022
| Acute myeloid leukaemia with defining genetic abnormalities |
| Acute promyelocytic leukaemia with PML::RARA fusion |
| Acute myeloid leukaemia with RUNX1::RUNX1T1 fusion |
| Acute myeloid leukaemia with CBFB::MYH11 fusion |
| Acute myeloid leukaemia with DEK::NUP214 fusion |
| Acute myeloid leukaemia with RBM15::MRTFA fusion |
| Acute myeloid leukaemia with BCR::ABL1 fusion |
| Acute myeloid leukaemia with KMT2A rearrangement |
| Acute myeloid leukaemia with MECOM rearrangement |
| Acute myeloid leukaemia with NUP98 rearrangement |
| Acute myeloid leukaemia with NPM1 mutation |
| Acute myeloid leukaemia with CEBPA mutation |
|
Acute myeloid leukaemia, myelodysplasia-related Defining cytogenetic abnormalities: |
|
Acute myeloid leukaemia with other defined genetic alterations Defining somatic abnormalities: |
| Acute myeloid leukaemia, defined by differentiation |
| Acute myeloid leukaemia with minimal differentiation |
| Acute myeloid leukaemia without maturation |
| Acute myeloid leukaemia with maturation |
| Acute basophilic leukemia |
| Acute myelomonocytic leukemia |
| Acute monocytic leukemia |
| Acute erythroid leukemia |
| Acute megakaryoblastic leukaemia |
ICC 2022
| Acute promyelocytic leukemia (APL) with t(15;17)(q24.1;q21.2)/PML::RARA ≥10% |
| APL with other RARA rearrangements* ≥10% |
| AML with t(8;21)(q22;q22.1)/RUNX1::RUNX1T1 ≥10% |
| AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22)/CBFB::MYH11 ≥10% |
| AML with t(9;11)(p21.3;q23.3)/MLLT3::KMT2A ≥10% |
| AML with other KMT2A rearrangements** ≥10% |
| AML with t(6;9)(p22.3;q34.1)/DEK::NUP214 ≥10% |
| AML with inv(3)(q21.3q26.2) or t(3;3)(q21.3;q26.2)/GATA2; MECOM(EVI1) ≥10% |
| AML with other MECOM rearrangements*** ≥10% |
| AML with other rare recurring translocations (see Supplemental Table 5) ≥10% |
| AML with t(9;22)(q34.1;q11.2)/BCR::ABL1‡ ≥20% |
| AML with mutated NPM1 ≥10% |
| AML with in-frame bZIP CEBPA mutations ≥10% |
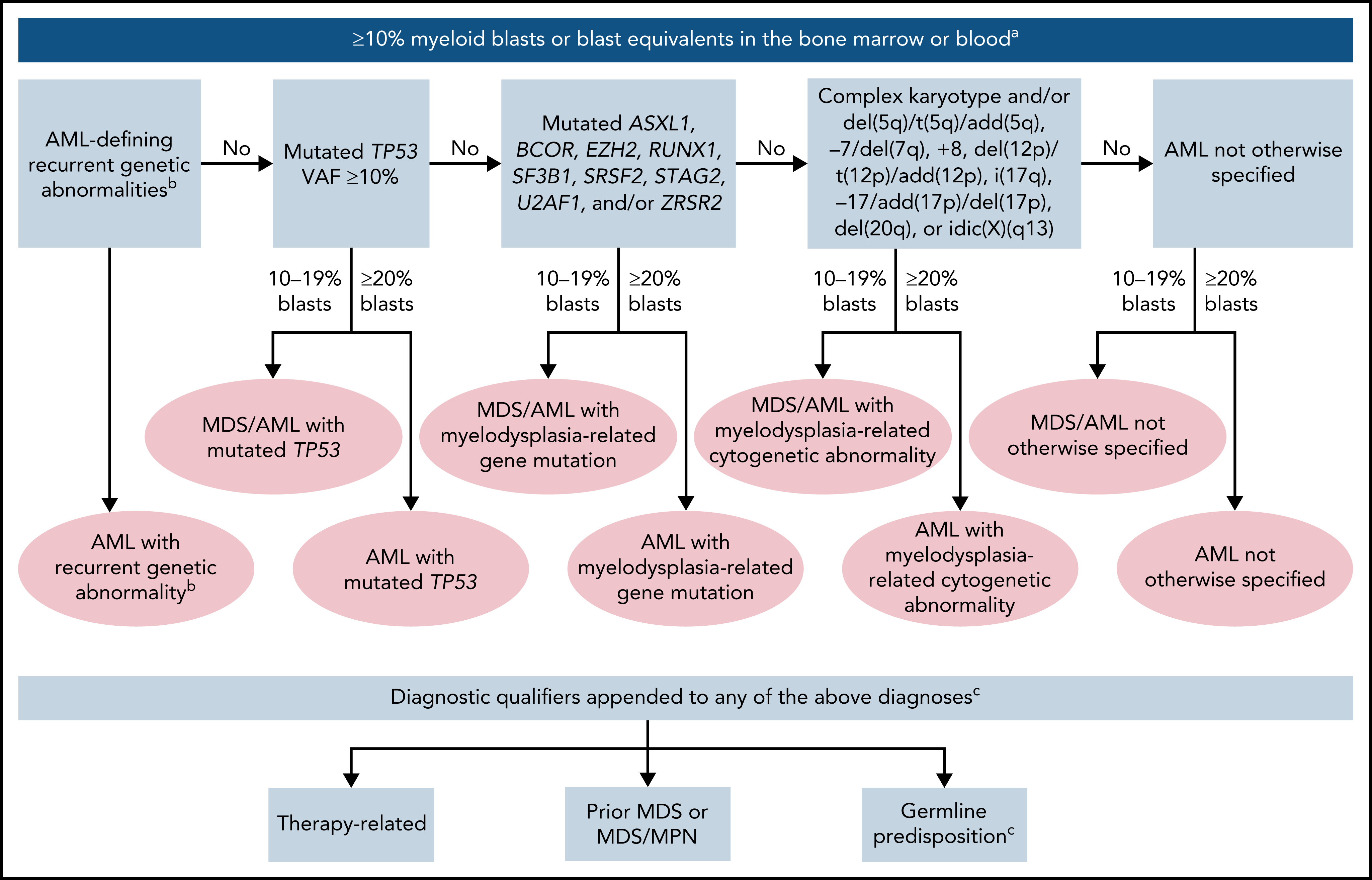
| AML and MDS/AML with mutated TP53† 10-19% (MDS/AML) and ≥20% (AML) |
| AML and MDS/AML with myelodysplasia-related gene mutations 10-19% (MDS/AML) and ≥20% (AML); Defined by mutations in ASXL1, BCOR, EZH2, RUNX1, SF3B1, SRSF2, STAG2, U2AF1, or ZRSR2 |
AML with myelodysplasia-related cytogenetic abnormalities 10-19% (MDS/AML) and ≥20% (AML)
|
| AML not otherwise specified (NOS) 10-19% (MDS/AML) and ≥20% (AML) Myeloid Sarcoma |
|
Footnotes: *Includes AMLs with: **Includes AMLs with: *** Includes AMLs with: ‡The category of MDS/AML will not be used for AML with BCR::ABL1 due to its overlap with progression of chronic myeloid leukemia, BCR::ABL1-positive Diagnostic qualifiers that should be used following a specific MDS, AML (or MDS/AML) diagnosis*
*Examples: Acute myeloid leukemia with myelodysplasia-related cytogenetic abnormality, therapy-related; acute myeloid leukemia with myelodysplasia-related gene mutation, progressed from myelodysplastic syndrome; AML with myelodysplasia-related gene mutation, germline RUNX1 mutation **lymphoblastic leukemia/lymphoma may also be therapy-related, and that association should also be noted in the diagnosis |
Hierarchical classification of the International Consensus Classification of AML
Immunofenotypering
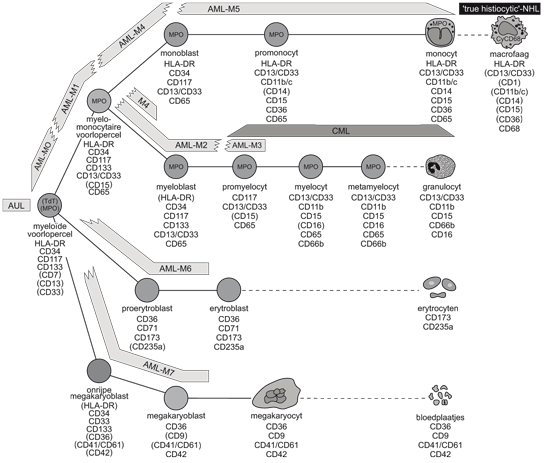
Op basis van het immunofenotype bij diagnose kan het Leukemia Associated Phenotype (LAP) worden vastgesteld en worden gebruikt voor de bepaling van minimale restziekte (MRD).
Onderzoek bij diagnose
- BSE, volledig bloedbeeld (Hb, Ht, erytrocyten, celindices, leukocyten, leukocyten differentiatie, trombocyten, reticulocyten).
- Chemie (ureum, kreatinine, urinezuur, Na, K, Ca, P, Mg, totaal eiwit, albumine, bilirubine, AF, γGT, ALAT, ASAT, LD, Fe, ferritine, transferrine, haptoglobine, vit. B12, foliumzuur, glucose).
- Bloedgroep, rhesus, directe Coombs.
- Diffuse intravasale stolling pakket.
- Beenmerg/Bloed: morfologie, immunologie, moleculaire diagnostiek (zoals in Hovon 132 studieprotocol ongeacht de leeftijd), cytogenetica, en indien informed consent cryopreservatie (biobank).
- Botbiopt.
- Aanvragen moleculaire diagnostiek op genetische predispositie indien geïndiceerd (zie ‘AML/MDS met germline predispositie’ tabel 2 en tabel 3). Diagnostiek wordt verricht op DNA uit bloed of beenmerg.
- Speeksel voor DNA indien indicatie germline onderzoek (zie ‘AML/MDS met germline predispositie’)
- Biobank, indien informed consent.
- Diagnostische liquorpunctie (1)(morfologie en immunologie) indien:
- neurologische symptomatologie
- leukocytose ≥ 40x 109/L
- extramedullaire ziekte (2)
- Virusserologie (HAV IgG, HBsAg, anti-HBc, anti-HCV, anti-HIV, anti-HTLV-1/2, IgG VZV, IgG CMV, IgG EBV VCA, IgG HSV-1/2).
- luesserologie.
- Urine algemeen onderzoek.
- Mantoux.
- ECG.
- HLA-typering patiënt klasse I (alle patiënten).
- HLA-typering klasse II (bij patiënten ≤ 70).
- X-thorax
- Echo-bovenbuik (lever, milt in cm).
- CT-thorax/abdomen op indicatie.
- Plaatsen Hickman-katheter.
- Sperma cryopreservatie op indicatie.
- Zwangerschaptest op indicatie.
- Counseling gynecologie op indicatie.
- NB bij morfologische verdenking op een Acute Promyelocyten Leukemie dient met spoed een PML/RAR kleuring met anti PML antilichaam te worden ingezet (morfologie lab).
Voetnoot (1) Een diagnostische liquorpunctie bij leukocytose en extramedullaire ziekte moet verricht worden na klaring van blasten uit het perifere bloed. In geval van neurologische symptomatologie wordt niet gewacht tot klaring van de blasten uit het perifeer bloed met het doen van een diagnostische liquorpunctie.
Voetnoot (2) Hepato/splenomegalie zal niet beschouwd worden als risicofactor voor CZS lokalisatie, gezien de relatief hoge prevalentie hiervan versus de lage prevalentie van CZS lokalisatie en de daaruit voortkomende lage diagnostische opbrengst.
NB: zie ook de specifieke studieprotocollen in verband met eventueel extra onderzoek.
AML/MDS met germline predispositie
Bij een deel van de AML patienten (schatting 5% maar toenemend in frequentie) is sprake van een aangeboren aanleg voor het ontwikkelen van myeloide maligniteiten. De onderliggende ‘germline’ mutaties kunnen overgeërfd zijn of ‘de novo’ ontstaan. De onderliggende mutaties leiden soms tot preleukemische syndromen (waaronder predispositie voor andere vormen van kanker), maar dit is niet altijd het geval.
Het is van belang de aanwezigheid van germline mutaties te identificeren omdat het belangrijke consequenties kan hebben voor de behandeling van de patiënt (indicatie en eligibility allogene SCT; donorkeuze; conditionering) maar ook voor genetische counseling en surveillance. Dit belang is onderstreept door vastlegging van ‘myeloid neoplasms with germ line predisposition’ in de WHO 2022 (zie tabel 1) en in de ELN richtlijnen (‘’Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel”)
Wanneer en hoe screening voor germline predispositie in een patiënt met AML/MDS?
Interdisciplinair overleg (tussen de afdelingen Hematologie en Klinische genetica van het Erasmus MC) heeft geleid tot een richtlijn voor screening op myeloide maligniteiten met germline predispositie die deels gebaseerd is op internationale richtlijnen. In tabel 2 wordt aangegeven wanneer een patiënt gescreend dient te worden voor de aanwezigheid van germline mutaties. De (familie)anamnese en gericht lichamelijk onderzoek (tabel 3) staan hierin centraal (naast de uitkomst van moleculair onderzoek). Figuur 1 laat het algoritme zien wat beschrijft op welke wijze screening plaats dient te vinden. Voor het inzetten van de screening dient de patiënt geïnformeerd te worden over het doel en de mogelijke consequenties van screening. In het uitkomstgesprek (gevoerd door hematoloog met kennis van leukemie-predispositie) is aandacht voor de betekenis van de test, de consequenties voor behandeling alsmede eventuele aanbevelingen voor orgaan/kanker screening/surveillance. Voor informatie over erfelijkheid en de eventuele screening van familieleden wordt patiënt verwezen naar de afdeling Klinische Genetica.
| Myeloid neoplasm classification | |
| Myeloid neoplasms with germ line predisposition without a preexisting disorder or organ dysfunction | |
| AML with germ line CEBPA mutation | |
| Myeloid neoplasms with germ line DDX41 mutation* | |
| Myeloid neoplasms with germ line predisposition and preexisting platelet disorders | |
| Myeloid neoplasms with germ line RUNX1 mutation* | |
| Myeloid neoplasms with germ line ANKRD26 mutation* | |
| Myeloid neoplasms with germ line ETV6 mutation* | |
| Myeloid neoplasms with germ line predisposition and other organ dysfunction | |
| Myeloid neoplasms with germ line GATA2 mutation | |
| Myeloid neoplasms associated with BM failure syndromes | |
| Myeloid neoplasms associated with telomere biology disorders | |
| JMML associated with neurofibromatosis, Noonan syndrome or Noonan syndrome-like disorders | |
| Myeloid neoplasms associated with Down syndrome* | |
* Lymphoid neoplasms also reported.
|
|
|
|
|
* 1e graad: kinderen, broers/zussen of ouder; 2e graads: oom/tante, neef/nicht, grootouders, kleinkinderen
|
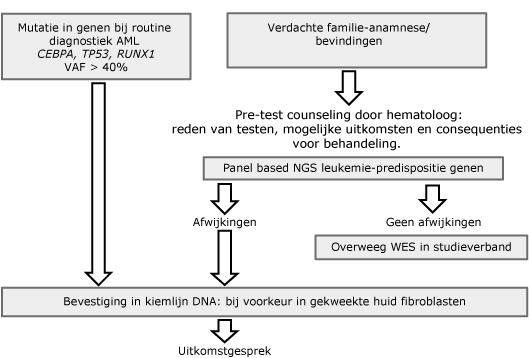
Figuur 1. Algoritme screening. NGS, next generation sequencing; WES, whole exome sequencing;
Behandeling
Hyperleukocytose
Definitie hyperleukocytose: WBC en/of blasten >100 x109/l.
Definitie symptomatische hyperleukocytose:
- neurologische symptomen, zoals verwardheid, slaperigheid, duizeligheid, hoofdpijn, delier, coma en parenchymale bloedingen.
- longcomplicaties, zoals hypoxemie, diffuse alveolaire bloedingen en respiratoire insufficiëntie.
Beleid:
- asymptomatisch: direct starten met hydroxycarbamide (minimaal 3 dd 1000 mg)
- symptomatisch: direct starten met intensieve chemotherapie teneinde snelle cytoreductie te bewerkstelligen.
Er is geen plaats meer voor leukaferese bij hyperleukocytose.
Initiële therapie AML
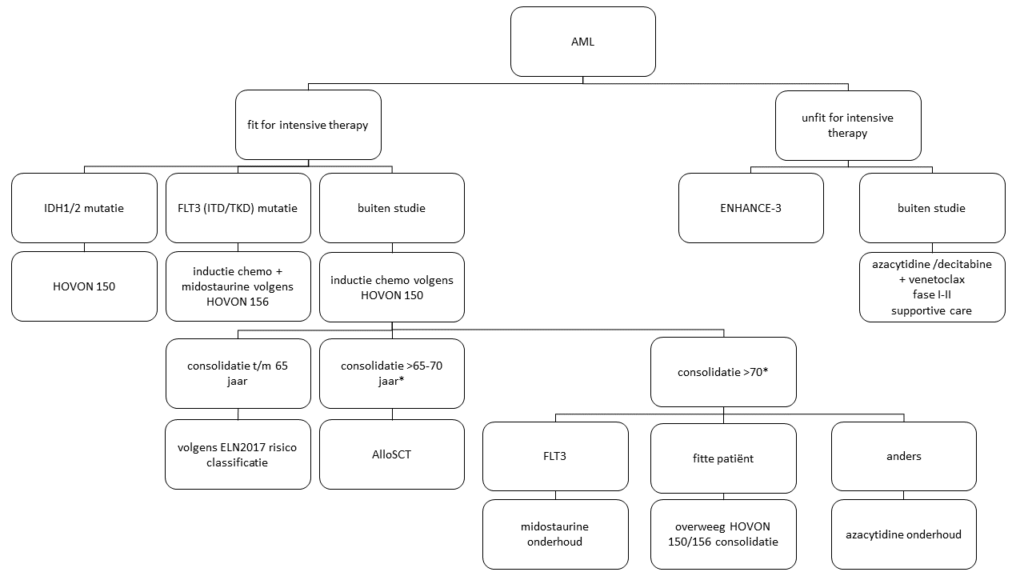
* Bij patiënten > 70 jaar kan onder voorwaarde ook een allogene stamceltransplantatie worden overwogen met name afhankelijk van fitheid en comorbiditeit.
Consolidatie
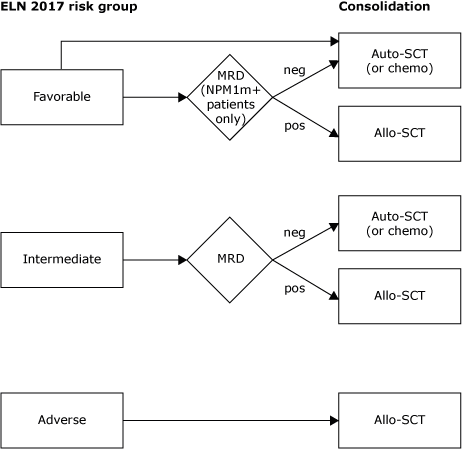
Voetnoten:
- MRD bepaling na remissie inductie cyclus 2
- MRD+ is gedefinieerd als moleculair (NPM1) en/of immunologische positiviteit
- Indien ELN 2017 “intermediate risk” maar zonder CRe: allo-SCT (onafhankelijk van MRD)
- Indien ELN 2017 “intermediate risk” maar leukocytose (>100): allo-SCT (onafhankelijk van MRD)
- Bij oudere patiënten (in het Erasmus MC gedefinieerd als >65-70 jaar) kan een allo-SCT worden overwogen ongeacht de ELN2017 risico-categorie gezien de in het algemeen slechte uitkomst van oudere patiënten in vergelijking met jongere patiënten. Bij deze patiënten moet het TRM risico worden meegewogen in de besluitvorming.
- Een eerder doorgemaakte MDS is geen onafhankelijke risicofactor voor slechtere uitkomst. EFS en OS lijkt ook in deze groep afhankelijk van cytogenetische (en moleculaire) kenmerken van de ziekte. Ook in deze groep van secundaire AML (evenals bij MDS-EB2 patiënten die intensieve remissie-inductie ondergaan) dient bepaling van de optimale consolidatie derhalve volgens dit algoritme te geschieden.
- In bovenstaand consolidatiealgoritme blijft vooralsnog gebruik gemaakt worden van de risicoclassificatie volgens ELN 2017; niet de risicoclassificatie volgen ELN 2022. Argumenten hiervoor zijn dat de waarde van MRD na inductiebehandeling, evenals de waarde van hyperleukocytose en het behalen van een CRe in “intermediate risk” patiënten vooralsnog onduidelijk zijn in de context van de ELN 2022 classificatie.
Azacitidine onderhoud
De prospectief gerandomiseerde QUAZAR studie heeft een significant overlevingsvoordeel laten zien van onderhoudsbehandeling met een orale formulering van azacitidine (Onureg, 300mg, 1dd gedurende 14 dagen in cycli van 28 dagen) bij patiënten die na inductiebehandeling niet in aanmerking kwamen voor een allogene SCT.
Op basis van deze studie adviseert de HOVON leukemie-werkgroep onderhoudsbehandeling met Onureg (300 mg, 1dd gedurende 14 dagen in cycli van 28 dagen) voor patiënten die met intensieve chemotherapie een complete remissie (CR/CRi) hebben bereikt maar niet in aanmerking komen voor consolidatiebehandeling met een hematopoiëtische stamceltransplantatie.
Midostaurin in de behandeling van FLT3 gemuteerde AML patiënten eligible voor intensieve chemotherapie:
Bij AML patiënten met een FLT3 mutatie (ITD of TKD) en eligible voor intensieve chemotherapie wordt de multi-kinase remmer midostaurin (Rydapt) toegevoegd aan de behandeling.
Midostaurin wordt conform de EMA registratie gegeven:
- direct aansluitend op de 2 remissie-inductiekuren, onafhankelijk van de leeftijd: dag 8 t/m 21, dosering 50 mg 2 d.d.
- direct aansluitend op de mitoxantrone/etoposide consolidatiekuur (indien gegeven): dag 8 t/m 21, dosering 50 mg 2 d.d.
- als onderhoudsbehandeling na consolidatie middels een derde kuur of autologe stamceltransplantatie. Midostaurin in de onderhoudsfase kan gestart worden na hematologisch herstel (ANC ≥ 1 x 109/L en trombocyten ≥ 50 x 109/L) en niet eerder dan 30 dagen na autologe stamceltransplantatie. Dosering is 50 mg 2 d.d., gedurende 1 jaar.
Richtlijnen m.b.t. gebruik en dosisaanpassingen van midostaurin in het Erasmus MC:
AML-studies
Zie KMS
HOVON 150: a phase 3, multicenter, double-blind, randomized, placebo-controlled study of ivosidenib or enasidenib in combination with induction therapy and consolidation therapy followed by maintenance therapy in patients with newly diagnosed acute myeloid leukemia or myelodysplastic syndrome with excess blasts-2, with an IDH1 or IDH2 mutation, respectively, eligible for intensive chemotherapy.
HOVON 156: a phase 3, multicenter, open-label, randomized, study of Gilteritinib versus Midostaurin in combination with induction and consolidation therapy followed by one-year maintenance in patients with newly diagnosed Acute Myeloid Leukemia (AML) or Myelodysplastic syndromes with excess blasts-2 (MDS-EB2) with FLT3 mutations eligible for intensive chemotherapy
ENHANCE-3 studie:
ENHANCE-3: A phase 3, randomized, double-blind, placebo-controlled study evaluating the safety and efficacy of Magrolimab versus placebo in combination with Venetoclax and Azacitidine in newly diagnosed, previously untreated patients with Acute Myeloid Leukemia who are ineligible for intensive chemotherapy
Behandeling van AML bij (oudere) patiënten die niet in aanmerking komen voor intensieve chemotherapie
Uitgangspunten
- Behandeling vindt zo veel mogelijk in studieverband plaats (zie boven onder “AML studies“)
- Bij een patiënt met een nieuw gediagnosticeerde AML die niet fit is voor intensieve chemotherapie maar wel behandeld wil worden, is er buiten studieverband de keuze uit hypomethylerende middelen (azacitidine of decitabine) of lage dosis cytarabine, al dan niet met toevoeging van de BCL2 remmer venetoclax. Monotherapie met hypomethylerende middelen of lage dosis cytarabine leveren overlevingswinst op ten opzichte van enkel ondersteunende behandeling (Fenaux P, J Clin Oncol. 2010;28(4):562-9; Kantarjian HM et al. J Clin Oncol. 2012; Burnett AK, et al. Cancer 2007).Toevoeging van venetoclax aan hypomethylerende therapie, zoals azacitidine, geeft een verbetering van respons en mediane overleving ten opzichte van monotherapie met hypomethylerende therapie (DiNardo et al, NEJM 2020). Hetzelfde geldt in iets mindere mate voor toevoeging van venetoxlax aan lage dosis cytarabine.
Op basis van bovenstaande gegevens is venetoclax in combinatie met hypomethylerende therapie de huidige standaardbehandeling bij niet fitte (oudere) patiënten met AML. Leidend in de keuze voor de optimale behandeling moet zijn de context van de patiënt, met name de performance status, niet noodzakelijkerwijs de absolute leeftijd.
Venetoclax + azacytidine (of venetoclax + decitabine)
| Middel | Dosering | Dagen |
| Azacitidine of: Decitabine |
75 mg/m2 sc
20 mg/m2 iv |
dag 1-7
dag 1-5 |
| Venetoclax | Ramp-up: 100 mg ∗ po 200 mg ∗ po 400 mg ∗ po |
dag 1 dag 2 dag 3-28 |
| ∗ = Dosisreductie tabel “Doseringsadvies ten aanzien van venetoclax in combinatie met CYP3A remmers” | ||
| Middel | Response status | Dosering | Dagen |
| Azacitidine of: Decitabine |
75 mg/m2 sc
20 mg/m2 iv |
dag 1-7
dag 1-5 |
|
| Venetoclax | <5% blasten en cellulariteit >20%** | 400 mg* | dag 1-21 of dag 1-14 *** |
| <5% blasten en cellulariteit <20%** | 400 mg* | dag 1-14 | |
| ≥5% blasten en cellulariteit >20% | 400 mg* | dag 1-28; start z.s.m. onafhankelijk van herstel | |
| ≥5% blasten en cellulariteit <20%**** | 400 mg* | dag 1-28 of dag 1-21 **** |
|
|
∗ Dosisreductie zie tabel “Doseringsadvies ten aanzien van venetoclax in combinatie met CYP3A remmers” |
|||
| Middel | Responsstatus | Dosering | Dagen |
| Azacitidine of: Decitabine |
75 mg/m2 s.c.
20 mg/m2 iv |
Dag 1-7
Dag 1-5 |
|
| Venetoclax | Bij voldoende hersteld bloedbeeld is een BM niet nodig | 400 mg* | Dag 1-14** |
| Bij progressieve verdenking progressieve ziekte, herhaal BM; als ≥5% blasten in BM | Stop behandeling | ||
|
* Dosisreductie zie tabel “Doseringsadvies ten aanzien van venetoclax in combinatie met CYP3A remmers” |
|||
Aandachtspunten:
- Aanpassing van de dosering venetoclax bij combinatie met CYP3A remmers, waarbij onderscheid gemaakt moet worden tussen matige CYP3A remmers (o.a. ciprofloxacine, fluconazol, isavuconazol, etc) en sterke CYP3A remmers (o.a. posaconazol, voriconazol, itraconazol, etc). Zie HOVON richtlijn AML 29-6-2021; pagina 85 (https://hovon.nl/_asset/_public/TreatmentGuidelines/TreatmentGuidelines_Leukemia/AML-richtlijn-versie-2021-06-29-definitief.pdf)
- In de praktijk is continue behandeling met venetoclax gedurende 28 dagen tijdens vervolgcycli doorgaans te toxisch en zijn aanpassingen in de duur van de behandeling noodzakelijk. Aanpassingen dienen plaats te vinden op basis van respons van ziekte (percentage blasten) in het beenmerg, beenmergcellulariteit en herstel van perifere bloedwaarden. Zie bovenstaande tabellen.
Alternatieve behandelopties:
- Monotherapie azacitidine 75 mg/m2 gedurende 7 opeenvolgende dagen per 28 dagen; responsbeoordeling na 4-6 kuren; behandeling tot progressie;
- Monotherapie decitabine 20 mg/m2 gedurende 10 opeenvolgende dagen per 28 dagen; duur 2e cyclus gestuurd door percentage blasten in het beenmerg op dag 28 (<5% dan verder met 5 dagen; ≥5% dan verder met 10 dagen; maximaal 3 kuren van 10 dagen; responsbeoordeling na 2-4 kuren; behandeling tot progressie);
- Lage dosis cytarabine (LDAC) 2 dd 20 mg sc, voor 10-14 dagen, elke 4-6 weken; behandeling tot progressie;
- Ondersteunende behandeling, al dan niet met hydroxycarbamide of 6-mercaptopurine, transfusies
Initiële therapie APL
Risicoclassificatie APL en behandeling
| Risicogroep: | Behandeling: |
|
Laag risico: |
Inductie: Consolidatie: Geen onderhoudsbehandeling klik hier voor het schema |
| Hoog risico: WBC > 10 x 109/L |
buiten studieverband: |
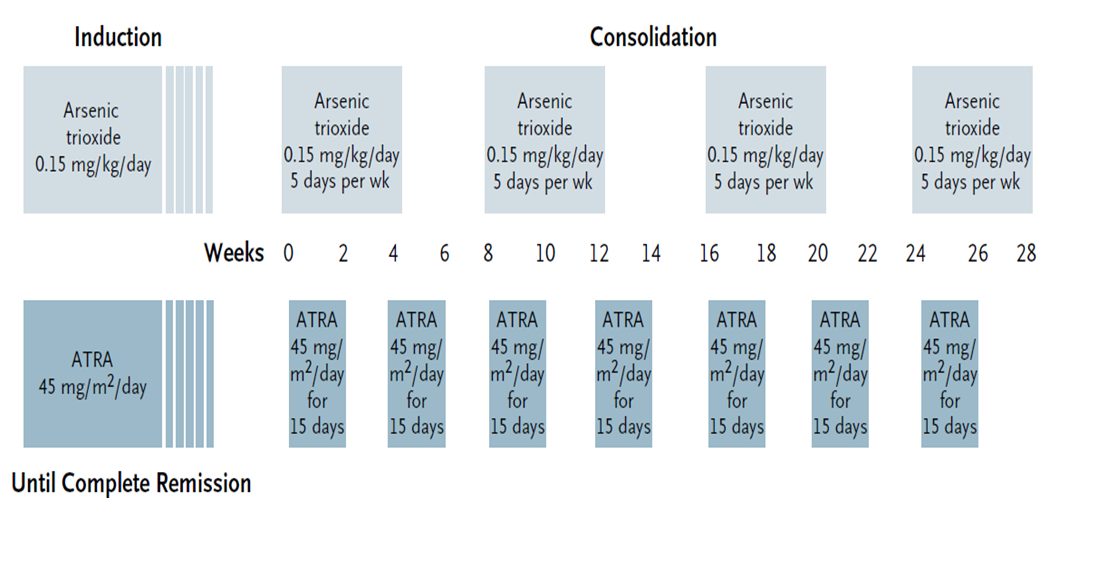{kind=link}
APL dosis aanpassingen (volgens de HOVON richtlijn)
- Voor kinderen/patiënten <20 jaar wordt de ATRA-dosering verlaagd naar 20 mg/m2
- Dexamethason (2 dd 5 mg; dag 1-15) wordt profylactisch gegeven bij een leukocyten aantal > 5 x 109/L ter voorkoming van het ATRA-syndroom (zie onderstaande); alternatief is 0.5 mg/kg prednisolon gedurende 3 weken.
- In geval chemotherapie wordt gegeven, dient bij patiënten > 70 jaar de dosering idarubicine gereduceerd te worden tot 3 dagen (dag 2, 4, 6) (i.p.v. 4 dagen (dag 2,4,6,8)) tijdens de inductie.
- Bij leukocytose: als leukocyten tussen 10-50 x109/L dan 4 dd 500 mg hydroxycarbamide toevoegen; als leukocyten >50 x109/L dan 4 dd 1000 mg hydroxycarbamide toevoegen. Hydroxycarbamide moet worden gestopt als leukocyten < 10 x109/L
Complicaties bij APL
Bloedingen of trombose bij APL
- De eerste 10 dagen het aantal trombocyten boven de 30×109/L en het Hb boven 5,5 mmol/L houden.
- Bij patiënten met een hoog risico op bloeding (> 70 jaar, leukocyten > 10×109/L, creatinine > 140 μmol/L) dienen de trombocyten boven 50×109/L gehouden te worden.
- Heparine en tranexaminezuur worden niet aanbevolen als profylaxe
- Het advies van experts is “liberaal” Omniplasma, plasma of cryoprecipitaat toe te dienen om het fibrinogeen boven 1-1,5 g/L te houden
- Gezien bloedingsrisico geen CVC plaatsen en geen leukaferese
Complicaties bij behandeling APL
APL differentiatiesyndroom (A-DS)
- Als een patiënt met APL die wordt behandeld met ATRA of ATO koorts, longinfiltraten, pleuravocht of pericard vocht ontwikkelt, dient men ervan uit te gaan (en zo te handelen) dat er sprake is van een
A-DS (ondanks de differentiaaldiagnose: pneumonie, decompensatie) en dient gestart te worden met steroïden (dexamethason 2 dd 10 mg). - Stijging van leukocyten bij ATRA/ATO combinatietherapie is op zich geen teken van A-DS, maar het optreden van een deel van deze symptomen (dus niet het complete scala van mogelijke symptomen) kan al wijzen op een A-DS. In principe dient de ATRA en/of ATO gecontinueerd te worden, tenzij de patiënt dusdanig ziek is dat hij/zij naar IC moet of als patiënt al 2 weken is behandeld met ATRA en/of ATO.
- Bij oplopend aantal leukocyten moet hydroxycarbamide worden toegevoegd (als leukocyten tussen 10-50 x109/L dan 4 dd 500 mg hydroxycarbamide; als leukocyten tussen >50 x109/L dan 4 dd 1000 mg hydroxycarbamide). Bij herstel van klachten kunnen de steroïden worden gestaakt en kan ATRA/ATO weer herstart worden (in 50% dosis, in een week op te bouwen tot 100% dosis).
- NB de aanwezigheid van meer dan 4 van de volgende kenmerken wordt beschouwd als ernstige A-DS: (onverklaarde) koorts, kortademigheid, pleura en/of pericardvocht, longinfiltraten, nierfalen, hypotensie en onverklaarde gewichtstoename meer dan 5 kg. Patiënten met 2 of 3 kenmerken worden geclassificeerd als matige A-DS (volgens Montesinos et al.).
Pseudotumor cerebri
Als de diagnose is gesteld (LP met vaststelling hoge liquor druk) dient ATRA te worden gestaakt en dexamethason (2 dd 5 mg) te worden gegeven. Ook hierbij kan ATRA weer hervat worden, eventueel gecombineerd met (profylactisch) dexamethason (2 dd 5 mg) na verbetering van de kliniek maar in lagere dosering
Bijwerkingen ATO
- QT-verlenging. In de ATRA/ATO NEJM studie: Een interval van > 450 ms voor mannen en > 460 ms voor vrouwen werd beschouwd als verlengd.
- Bij QTc boven deze waarden werd de toediening van ATO en ook andere medicatie met invloed op QTc onderbroken en elektrolyten werden gecorrigeerd (indien nodig).
- Bij normalisatie van QTc werd ATO hervat in dosering van 0,075 mg/kg (50%) .Als in de eerste week dan geen verdere verlenging optrad werd de ATO stapsgewijs opgehoogd naar 0,11 mg/kg. Als dit dosisniveau ook gedurende een week geen QTc verlenging gaf, werd de dosering ATO verder opgehoogd tot de volledige dosis.
- Derhalve is het uitdrukkelijk advies om het ECG minimaal 1x per week te verrichten en in geval van tekenen van QT verlenging 2-3 x per week. Aandacht voor normale waarden van elektrolyten.
- Hepatotoxiciteit. Bij graad 3-4 CTCAE hepatotoxiciteit (bilirubine en/of ASAT en/of AF > 5x ULN) is het advies om tijdelijk ATRA en/of ATO te staken. ATRA en/of ATO kunnen herstart worden in 50% dosering als de leverwaarden zijn gedaald naar < 4x ULN. Als dit gedurende een week goed gaat, de dosering ATRA en/of ATO hervatten op 100%. Bij terugkeer van hepatotoxiciteit de middelen blijvend stoppen.
APL respons evaluaties
- Eerste evaluatie beenmergaspiraat wordt verricht op dag 28 van de inductiekuur. Indien nog geen morfologische complete remissie is bereikt dan wordt wekelijks een beenmergaspiraat herhaald tot bereiken CR (remissie wordt altijd bereikt). Bij remissie (< 5% myeloblasten en geen abnormale promyelocyten) staak ATO/ATRA. Bevestig remissie na hematologisch herstel (N > 1 en Tr > 100).
- Follow-up parameters: De complete remissie wordt bepaald met een PCR op PML-RARA, het transcript van het fusie-gen. Deze PCR heeft een gevoeligheid van 10-4 – 10-5 en dient op het beenmerg negatief te zijn na het afsluiten van de inductie- en consolidatiebehandeling (dus voor het starten van de onderhoudsfase).
- Een blijvend positieve PCR voor PML-RARA impliceert dat de leukemie niet in een remissie is gekomen en vereist aanvullende behandeling. Een allogene HCT moet dan sterk worden overwogen. Verder kan de PCR (bijv. 3-maandelijks) (om PML-RARA te detecteren) herhaald worden na behandeling van de high-risk APL groep gedurende de eerste 2 jaar na behandeling.
Therapie bij refractaire ziekte en na recidief
Beslissen over keuze van therapie afhankelijk van risicofactoren (leeftijd, risicoindeling leukemie, voorafgaande transplantatie, duur van de remissie voorafgaande aan het recidief, comorbiditeit, eerdere complicaties) en van moleculaire kenmerken (waaronder eventuele aanwezigheid FLT3-mutatie bij recidief). Bij een recidief AML dient opnieuw moleculair-genetisch onderzoek verricht te worden. In het algemeen is de prognose van een recidief AML beperkt.
De therapeutische mogelijkheden zijn:
- reïnductie chemotherapie gevolgd door allogene stamceltransplantatie of DLI;
- indien FLT3-mutatie (ITD of TKD): gilteritinib gevolgd door allogene stamceltransplantatie of DLI;
- donor lymfocyten infusie (DLI) bij recidief in een tevoren getransplanteerde patiënt;
- fase I-II studies zie KMS;
- venetoclax in combinatie met azacitidine (of decitabine of lage dosis cytarabine) kan overwogen worden maar is niet geregistreerd voor deze indicatie;
- palliatieve behandeling
Recidief na een eerdere complete remissie (zonder allogene SCT)
Bij recidief van AML (waarbij in CR1 geen alloSCT): opnieuw remissie inductie behandeling en in geval van een nieuwe CR – alloSCT mits eligible.
Opties voor remissie-inductiebehandeling, afhankelijk van ziekte- en patientgerelateerde factoren:
- inductie chemotherapie: Cytarabine 1500 mg/m2 a 12 uur op dag 1, 3, 5, 7 en Daunorubicine 90 mg/m2 op dag 1 t/m 3
Let op cumulatieve anthracycline dosering ( https://www.vademecumhematologie.nl/berekeningen/cumulatieve-antracycline-dosering/)
Aanvullend geldt: bij risicofactoren voor antracycline-geinduceerde cardiotoxiciteit overweeg daunorubicine 60 mg/m2 op dag 1 t/m 3. Risicofactoren zijn o.a.:- Leeftijd > 65 jaar
- Eerder cardiovasculair event
- Risicofactoren voor cardiovasculair event (roken, DM, hypertensie)
- LVEF < 50% (55%)
- Radiotherapie met exposure hart
- Chronische nierziekte
- Indien bij refractaire ziekte/recidief een FLT3-mutatie (ITD of TKD) aantoonbaar is: gilteritinib monotherapie. Dit kan ook effectief zijn na eerdere behandeling met de FLT3-remmer midostaurine in de eerste lijn.
- venetoclax in combinatie met azacitidine (of decitabine of lage dosis cytarabine) kan overwogen worden, met name bij vroeg recidief, maar is niet geregistreerd voor deze indicatie; een eventuele aanvraag voor toestemming van gebruik dient vooraf individueel afgewogen te worden.
In overige gevallen van recidief: onderzoek de mogelijkheid van Fase I-II studies.
Indien er geen passende Fase I-II studie beschikbaar is of eventueel patiënt de mogelijkheid van Fase I-II studie afwijst: palliatieve behandeling
Recidief na allogene SCT
- Hernieuwde remissie-inductie en indien morfologisch CR gevolgd door DLI, wanneer aan de volgende voorwaarden wordt voldaan:
- geen actieve GvHD
- in verleden geen acute GVHD III-IV of chr. ext. GvHD gehad
- relapse niet binnen 6 mnd na alloSCT.
-
Opties voor remissie-inductie: zie boven (inductie chemotherapie, gilteritinib of venetoclax/azacitidine, afhankelijk van patiëntspecifieke factoren)
- Onderzoek de mogelijkheid van Fase I-II studie.
Primair refractaire ziekte
In principe gelden dezelfde overwegingen als boven genoemd bij recidief.
In een geselecteerde groep patiënten kan overwogen worden om door te gaan naar allogene stamceltransplantatie ondanks het niet behalen van een complete remissie. Hierbij kan gebruikt gemaakt worden van onderstaande risicoscore (Todisco et al, Bone Marrow Transplantation (2017) 52, 955-961), waarbij deze optie alleen overwogen kan worden bij patiënten met score 0 (0 of 1 risicofactoren).
| Variabele | Score |
| Aantal cycli chemotherapie | |
| ≤ 2 | 0 |
| > 2 | 1 |
| Blast infiltratie | |
| Beenmerg < 25%; geen in perifeer bloed | 0 |
| Beenmerg ≥ 25% of blasten in perifeer bloed | 1 |
| Leeftijd | |
| ≤ 60 jaar | 0 |
| > 60 jaar | 1 |
| Cytogenetica/moleculair | |
| Favorable/Intermediate I | 0 |
| Intermediate II/Adverse | 1 |
| Score | Risicofactoren | HR (95% CI) | 3 jaar OS |
| 0 | 0-1 | 32% | |
| 1 | 2 | 1.73 (1.13-2.63) | 10% |
| 2 | 3-4 | 2.62 (1.68-4.10) | 3% (2 jaar) |
Recidief APL
Behandeling met arseentrioxide (ATO)
- Inductiekuur 0.15 mg/kg/dag, 7 dagen per week, i.v. 1-2 uurs infuus tot morfologisch CR, met maximum van 60 dagen.
(CR = neutro’s >1500/µl, platelets >100000/µl in perifeer bloed, blasten + promyelocyten <5% in BM. (BM aspiraat verrichten als criteria voor CR in perifeer bloed zijn bereikt, op dg 60 van ATO, of eerder als nodig).
Indien na 60 dgn geen morfologisch CR: combinatietherapie van ATO, ATRA en/of chemotherapie (bijv hoge dosis cytarabine). - Consolidatiekuur 0.15 mg/kg/dag i.v., 5 dgn/wk, 5 wk (start na 4 weken na einde inductie-kuur).
- Vervolgbehandeling afhankelijk van PCR na consolidatiekuur.
| PCR positief: | 2e consolidatiekuur. Nadien PCR positief: overweeg eerst combinatie van ATO, ATRA en/of chemotherapie alvorens verder te gaan naar allogene PSCT (bij lft <40 jr) |
| Nadien PCR negatief: zie verder | |
| PCR negatief: | Autologe PSCT |
Respons criteria
De tabel met respons criteria is ontleend aan de publicatie van Döhner H, et al. “Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel”. Blood, 2017:129:424-447
| Category | Definition | Comment |
| Response: | ||
| CR without minimal residual disease CRMRD- | If studied pretreatment, CR with negativity for a genetic marker by RT-qPCR, or CR with negativity by MFC | Sensitivities vary by marker tested, and by method used; therefore, test used and sensitivity of the assay should be reported; analyses should be done in experienced laboratories (centralized diagnostics) |
| Complete remission (CR) | Bone marrow blasts <5%; absence of circulating blasts and blasts with Auer rods; absence of extramedullary disease; ANC ≥1.0 × 109/L (1000/µL); platelet count ≥100 × 109/L (100 000/µL) | MRD+ or unknown |
| CR with incomplete hematologic recovery (CRi) | All CR criteria except for residual neutropenia (<1.0 × 109/L [1000/µL]) or thrombocytopenia (<100 × 109/L [100 000/µL]) | |
| Morphologic leukemia-free state (MLFS) | Bone marrow blasts <5%; absence of blasts with Auer rods; absence of extramedullary disease; no hematologic recovery required | Marrow should not merely be “aplastic”; at least 200 cells should be enumerated or cellularity should be at least 10% |
| Partial remission (PR) | All hematologic criteria of CR; decrease of bone marrow blast percentage to 5% to 25%; and decrease of pretreatment bone marrow blast percentage by at least 50% | Especially important in the context of phase 1-2 clinical trials |
| Treatment failure: | ||
| Primary refractory disease | No CR or CRi after 2 courses of intensive induction treatment; excluding patients with death in aplasia or death due to indeterminate cause | Regimens containing higher doses of cytarabine (see Table 8) are generally considered as the best option for patients not responding to a first cycle of 7+3; the likelihood of responding to such regimens is lower after failure of a first |
| Death in aplasia | Deaths occurring ≥7 d following completion of initial treatment while cytopenic; with an aplastic or hypoplastic bone marrow obtained within 7 d of death, without evidence of persistent leukemia | |
| Death from indeterminate cause | Deaths occurring before completion of therapy, or <7 d following its completion; or deaths occurring ≥7 d following completion of initial therapy with no blasts in the blood, but no bone marrow examination available | |
| Response criteria for clinical trials only: | ||
| Stable disease | Absence of CRMRD−, CR, CRi, PR, MLFS; and criteria for PD not met | Period of stable disease should last at least 3 mo |
| Progressive disease (PD)*,† |
Evidence for an increase in bone marrow blast percentage and/or increase of absolute blast counts in the blood:
|
Category mainly applies for older patient given low-intensity or single-agent “targeted therapies” in clinical trials Some protocols may require blast increase in 2 consecutive marrow assessments at least 4 wk apart; the date of progression should then be defined as of the first observation date Some protocols may allow transient addition of hydroxyurea to lower blast counts “Progressive disease” is usually accompanied by a decline in ANC and platelets and increased transfusion requirement and decline in performance status or increase in symptoms |
| Relapse: | ||
| Hematologic relapse (after CRMRD−, CR, CRi) | Bone marrow blasts ≥5%; or reappearance of blasts in the blood; or development of extramedullary disease | |
| Molecular relapse (after CRMRD−) | If studied pretreatment, reoccurrence of MRD as assessed by RT-qPCR or by MFC | Test applied, sensitivity of the assay, and cutoff values used must be reported; analyses should be done in experienced laboratories (centralized diagnostics) |
|
ANC, absolute neutrophil count; IDH, isocitrate dehydrogenase; MLFS, morphologic leukemia-free state; WBC, white blood cell.
*The authors acknowledge that this new provisional category is arbitrarily defined; the category aims at harmonizing the various definitions used in different clinical trials. †Certain targeted therapies, for example, those inhibiting mutant IDH proteins, may cause a differentiation syndrome, that is, a transient increase in the percentage of bone marrow blasts and an absolute increase in blood blasts; in the setting of therapy with such compounds, an increase in blasts may not necessarily indicate PD.
|
||
| Category | Definition | Comment |
| Response: | ||
| Complete remission (CR)a,b,c | Bone marrow blasts <5%; absence of circulating blasts or blasts with Auer rods; absence of extramedullary disease; ANC ≥1.0 x 109/L (1,000/μL); platelet count ≥100 x 109/L (100,000/μL) | |
| CR with partial hematologic recovery (CRh)a,b,c | ANC ≥0.5 x 109/L (500/μL) and platelet count ≥50 x 109/L (50,000/μL), otherwise all other CR criteria met | If CRh used, CRi should only include patients not meeting the definition of CRh |
|
CR with incomplete hematologic recovery (CRi)a,b,c |
All CR criteria except for residual neutropenia <1.0 x 109/L (1.000/μL) or thrombocytopenia <100 x 109/L (100,000/μL) | |
| Morphologic leukemia-free state (MLFS) | Bone marrow blasts <5%; absence of blasts with Auer rods; absence of circulating blasts; absence of extramedullary disease; no hematologic recovery required | Marrow should not merely be “aplastic”; bone marrow spicules should be present; at least 200 cells should be enumerated in the aspirate or cellularity should be at least 10% in the biopsy. Mainly used in the context of phase 1-2 clinical trials |
| Partial remission (PR) |
All hematologic criteria of CR; decrease of bone marrow blast percentage to 5% to 25%; and decrease of pre-treatment bone marrow blast percentage by at least 50% |
Mainly used in the context of phase 1-2 clinical trials |
| No response |
Patients evaluable for response but not meeting the criteria for CR, CRh, CRi, MLFS or PR are categorized as having no response prior to the response landmark. Patients failing to achieve response by the designated landmark are designated as having refractory disease |
|
| Non-evaluable for response |
Non-evaluable for response will include patients lacking an adequate bone marrow response evaluation. This category will include patients with early death, withdrawal prior to response assessment, or a technically suboptimal bone marrow sample precluding assessment |
|
| Response ((if including assessment of MRD)d | ||
|
CR, CRh or CRi without MRDc (CRMRD-, CRhMRD- or CRiMRD-) |
CR, CRh or CRi with MRD below a defined threshold for a genetic marker by qPCR, or by MFC. Response with MRD detection at low-level (CRMRD-LL) is included in this category of CR, CRh or CRi without MRD. CRMRD-LL is currently only defined for NPM1-mutant and CBF-AML |
Sensitivities vary by marker tested, and by method used; therefore, test used, tissue source and minimum assay sensitivity for evaluability should be reported; analyses should be done in experienced laboratories (centralized diagnostics) |
| Treatment failure | ||
| Refractory disease | No CR, CRh or CRi at the response landmark, ie, after 2 courses of intensive induction treatment or a defined landmark, eg, 180 days after commencing less-intensive therapy | Patients not responding to a first cycle of 7+3 should be considered for a regimen containing higher doses of cytarabine |
| Relapsed disease (after CR, CRh or CRi) | Bone marrow blasts ≥5%; or reappearance of blasts in the blood in at least 2 peripheral blood samples at least one week | |
| ANC, absolute neutrophil count; CBF, core-binding factor; MFC, multi-parameter flow cytometry; MRD; measurable residual disease; NGS, next-generation sequencing; qPCR, quantitative polymerase chain reaction; VAF, variant allele frequency a) To recognize the potential for continuing improvements in blood counts after myelosuppressive therapy, response definitions for patients with marrow blast clearance (<5%) may be adjusted to reflect the best hematologic response achieved prior to commencement of the next treatment cycle. Aspirate reports that include MLFS, CRh, or CRi should note the potential for post-marrow blood counts to alter the final response designation. Patients should not have received G-CSF, nor platelet transfusions within 7 days prior to hematologic response determination. b) For patients with CR, CRh or CRi, the presence of a low percentage of circulating blasts in the blood may represent a regenerating marrow and should not be interpreted as persistent disease. In such cases the blasts generally disappear within a week. c) A response landmark for CR, CRh or CRi should be stated, eg, after two cycles of intensive therapy; this landmark may be longer for non-intensive based treatment options, eg,180 days. d) MFC-MRD positivity is defined as 0.1% of CD45 expressing cells with the target immunophenotype. MRD test positivity by qPCR is defined as cycling threshold (Ct) <40 and is negative if Ct 40 in 2 of 3 replicates. In NPM1-mutated and CBF-AML, CR with molecular MRD detectable at low-level (CRMRD-LL) defined as <2% is designated as negative for MRD, because when measured at the end of consolidation treatment, is associated with a very low relapse rate. NB) zie de blokjes bij de Ct waarde – |
||