
Indeling WHO-classificatie (2016):
- Chronische myeloide leukemie, BCR-ABL1 positief (CML).
- De klassieke myeloproliferatieve aandoeningen:
- polycythemia vera (PV);
- essentiële trombocytose (ET);
- primaire myelofibrose (PMF):
- primaire myelofibrose, prefibrotisch/vroeg stadium
- primaire myelofibrose, overt fibrotisch stadium
- Een restgroep van zeldzaam voorkomende myeloproliferatieve ziektes:
- chronische neutrofielen leukemie (CNL);
- chronische eosinofielen leukemie, niet nader gespecificeerd (CEL, nos);
- niet nader te classificeren myeloproliferatieve aandoening (MPN, u).
Polycythemia vera (PV)
Diagnostische criteria
Diagnostische WHO-criteria (2016)
|
Major criteria:
|
|
Minor criterium:
|
|
Diagnose PV:
|
|
* = Major criterium 2 is niet noodzakelijk indien er sprake is van een absolute erytrocytose (Hb >11,6 mmol/l en/of H t>0.56 l/l voor mannen; Hb >10,3 mmol/l en/of Ht >0.50 l/l voor vrouwen) en voldaan wordt aan de andere criteria |
Onderzoek
- Anamnese met aandacht voor algemene klachten, constitutionele symptomen (bij voorkeur met Myeloproliferative Neoplasm-Symptom Assessment Form (MPN-SAF vragenlijsten), bloedingsneiging, cardiovasculaire risico factoren en jicht
- Lichamelijk onderzoek: bloeddruk, miltgrootte
- Bloedbeeld (inclusief leukocyten differentiatie), reticulocyten, kreatinine, leverenzymen, LDH, urinezuur, ferritine, glucose en cholesterol/triglyceriden
- Mutatie bepaling: JAK2V617F mutatie, BCR-ABL1 mutatie (ter uitsluiting CML) en bij een negatieve testuitslag JAK2 exon12 mutatie
- Op indicatie:
- Beenmergbiopt en morfologie
- Erytropoëtine spiegel, voor start aderlatingen
- Echo milt bij twijfel over splenomegalie
- In geval van verhoogde bloedingsneiging en/of indien trombocytose voorafgaand aan ingrepen met bloedingsrisico: sluit verworven von Willebrand type II uit middels bepaling van von Willebrand ristocetine activiteit en antigeen (verstoorde ratio (0,7), met verminderde activiteit)
Stroomdiagram diagnostiek erytrocytose
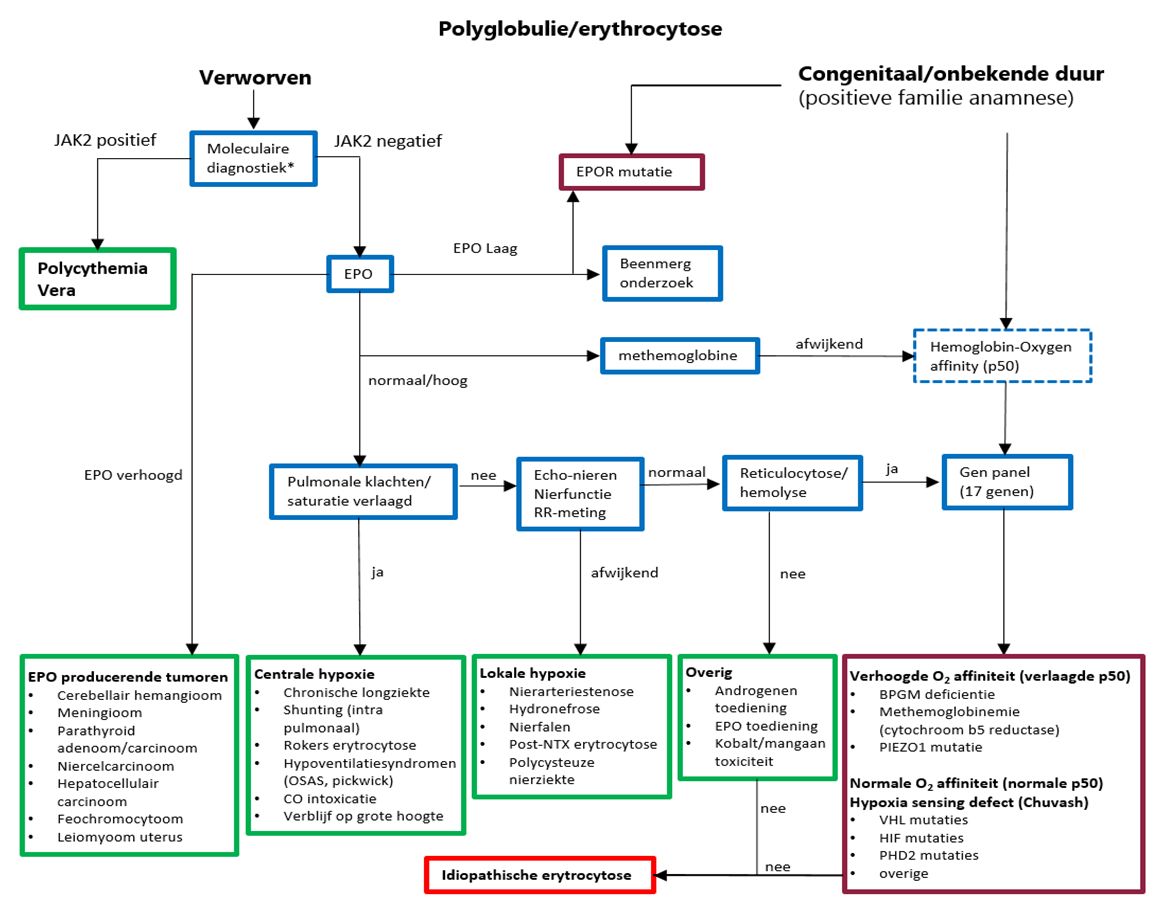
Oorzaken van primaire en secundaire erytrocytose
| Primaire erytrocytose: | ||
| Congenitaal | ||
| Erytropoëtine receptor (EpoR) mutaties Lymphocyte adaptor protein (LNK) mutaties (SH2B3 gen) |
||
| Verworven | ||
| Polycythemia Vera (PV) | ||
| Secundaire erytrocytose: | ||
| Congenitaal | ||
| Defecten van de ‘oxygen sensing pathway’ |
|
|
| Andere congenitale defecten |
|
|
| Verworven | ||
| Centrale hypoxie |
|
|
| Lokale hypoxie |
|
|
| Pathologische erytropoëtine productie |
|
|
| Overig |
|
|
Risico classificatie
| Hoog risico | aanwezigheid van 1 of meerdere risicofactoren |
| Standaard risico | geen hoog risico factoren |
| Risico factoren | leeftijd > 60 jaar eerdere trombo-embolische complicatie (leukocytose > 15 x 109/L) |
Behandeling
Algemeen
- behandeling hoog uraatgehalte (allopurinol 1 dd 300 mg);
- pruritus: antihistaminica, H2-antagonisten, cetirizine (Zyrtec®);
- Ascal 1 dd 80 mg of carbasalaatcalcium 1 dd 100 mg indien er geen contra-indicaties zijn.
Overwogen kan worden te kiezen voor een avond dosering i.p.v. een ochtend dosering.
Specifiek:
Polycythemische fase
- Flebotomie: wekelijks 250 – 500 ml per keer totdat Ht <0.45, daarna op geleid van de hematocriet.
- Cytoreductieve therapie:
- Indicaties voor cytoreductieve therapie bij hoog risico PV
- leeftijd > 60 jaar
- (doorgemaakte) trombo-embolische complicatie
- (leukocytose >15 x 109/L)
- Overige indicaties voor cytoreductieve therapie
- trombocyten >1500 x 109/l of verworven vWD, streef verlagen trombocyten en/of remissie VvWD
- Symptomatische splenomegalie
- Te frequente flebotomieën, streef acceptabel flebotomie interval
- Slecht verdragen flebotomieën
- Progressieve myeloproliferatie bij hoog risico PV, streef verlagen trombocyten <400 x109/L en/of leukocyten <15 x109/l
- N.B. Indien de Ht controle middels flebotomieën plaats vindt en geen ander indicatie behoudens de leeftijd bestaat voor het starten van cytoreductieve therapie, kan overwogen worden het starten van cytoreductieve therapie uit te stellen (expert opinion).
- Indicaties voor cytoreductieve therapie bij hoog risico PV
Behandelopties
- 1e lijn:
- Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Gepegyleerd interferon α 2a (Pegasys®), start dosering 45-90 microgram/week s.c.
- 2e lijn en verder:
- Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Gepegyleerd interferon α 2a (Pegasys®), start dosering 90 microgram/week s.c.
- In de loop van 2022 zal een andere gepegyleerde vorm van interferon op de Nederlands markt geintroduceerd worden: Ropegferon. Dit geneesmiddel is alleen geregistreerd voor PV behandeling!
- Ruxolitinib, start dosering 2 dd 10 mg, dosering aan te passen op therapeutisch effect en trombocyten aantal tenminste > 50-100 x 109/L (streefwaarde > 100 x 109/L). Aanpassen dosering met 1-2 dd 5 mg ophogen per 2 a 3 weken tot een max. dosering van 2 dd 25 mg afhankelijk van het bereikte effect.
- Indicatie algemeen: onvoldoende effect op conventionele cytoreductieve therapie
- Indicatie specifiek:
- Hydroxycarbamide resistentie (volgens ELN criteria):
- Het niet bereiken van een adequaat therapeutisch effect bij een dosering van ≥2 g/dag gedurende 12 weken therapie, OF indien het beoogde therapeutisch effect niet bereikt wordt bij een maximaal te tolereren dosering van <2 gram/dag
Onder een inadequaat therapeutisch effect wordt een of meer van de volgende waarnemingen verstaan:
- onvoldoende reductie op trombocyten/leukocyten aantal
- onvoldoende effect op symptomatische splenomegalie
- Hydroxycarbamide intolerantie
- Invaliderende jeuk die niet of onvoldoende reageren op een eerdere lijn(en) van therapie
- Onvoldoende Ht beheersing d.m.v. flebotomie en/of cytoreductieve therapie
- Ernstige vermoeidheid die gerelateerd is aan PV
- Het ontstaan van ulcera aan de benen of ander onacceptabele toxiciteit (gedefinieerd als graad 3-4 toxiciteit (CTC) zoals mucocutane manifestaties, gastro-intestinale symptomen, pneumonitis, of koorts
- Hydroxycarbamide resistentie (volgens ELN criteria):
- Anagrelide (overweeg bij trombocytose). Startdosering 2 dd 0,5 mg, iedere week te verhogen met een 0,5 mg/dag extra op geleide van het trombocyten aantal. Maximaal 2 dd 5 mg
- Combinatie van behandelingen
- Busulfan, start dosering 2-4 mg/dag. (CAVE langdurige cytopenie: controle bloedbeeld en cave leukemogeniciteit)
- Melfalan, start dosering 2 mg 3 x/week. (CAVE cytopenie: controle bloedbeeld en cave leukemogeniciteit)
- Fosfor-32 (32P), dosering 3mCI i.v, zonodig na 3 maanden herhalen (CAVE leukemogeniciteit)
Indien de diagnose PV niet gesteld wordt, is er geen indicatie voor trombocyten aggregatieremming of nastreven van een Ht <0,45 l/l. Uit reologisch oogpunt kan overwogen worden te streven naar een Ht <0.55 l/l indien men problemen verwacht bij oplopende viscositeit bij hogere Ht waardes. In geval van trombo-embolische complicaties in de voorgeschiedenis kan overwogen worden een streefwaarde voor het Ht <0,45 l/l aan te houden.
Post PV-MF/Spent-phase
Zie onderdeel Primaire Myelofibrose
Essentiële trombocytemie (ET)
Diagnostische criteria
Voor het stellen van de diagnose ET kan gebruik gemaakt worden van de WHO (2016) criteria.
Diagnostische WHO-criteria (2016)
| Major criteria: |
|
| Minor criterium: |
|
|
Diagnose ET:
|
Onderzoek
- Anamnese met aandacht voor algemene klachten, constitutionele symptomen (bij voorkeur met met Myeloproliferative Neoplasm-Symptom Assessment Form (MPN-SAF) vragenlijsten), bloedingsneiging en cardiovasculaire risico factoren.
- Lichamelijk onderzoek: bloeddruk, miltgrootte
- Bloedbeeld (inclusief leukocyten differentiatie), reticulocyten, kreatinine, leverenzymen, LDH, urinezuur, ferritine, CRP, glucose, cholesterol/triglyceriden
- Mutatie bepaling: JAK2V617F, BCR-ABL1 mutatie (ter uitsluiting CML) en bij een negatieve testuitslag CALR en MPL mutatie
- Beenmergbiopt
- Op indicatie:
- Echo milt bij twijfel over splenomegalie
- In geval van verhoogde bloedingsneiging en/of indien trombocytose voorafgaand aan ingrepen met bloedingsrisico: sluit verworven von Willebrand type II uit middels bepaling van von Willebrand ristocetine activiteit en antigeen (verstoorde ratio (0,7), met verminderde activiteit)
- Cytogenetisch onderzoek (bijvoorbeeld ter uitsluiting MDS) indien geen mutaties aangetoond
Behandeling
Risicogroepen
Afhankelijk van leeftijd, JAK2 mutatie status, cardiovasculair risicoprofiel en eventueel eerder opgetreden trombo-embolische complicaties worden ET patiënten in verschillende risico groepen ingedeeld.
IPSET criteria voor trombose risico
| Risico factor | HR | score |
| Leeftijd > 60 jaar | 1.50 | 1 |
| Cardiovasculaire risicofactoren | 1.56 | 1 |
| Eerdere trombo-embolische complicatie | 1.93 | 2 |
| JAK2v617F mutatie | 2.04 | 2 |
| Laag risico score: 0-1 Intermediair risico: 2 Hoog risico: ≥ 3 |
||
Behandeling
Laag risico patiënten (volgens IPSET score)
- Ascal 100 mg/dag. Overwogen kan worden, afhankelijk van het risico op bloedingen, geen trombocytenaggregatieremmers te starten.
- Op indicatie: behandelen vasculair risico profiel.
Intermediair/Hoog risico patiënten (volgens IPSET score)
- Ascal 100 mg/dag. Overwogen kan worden te kiezen voor een avond dosering i.p.v. een ochtend dosering
- Op indicatie: behandelen vasculair risico profiel.
- Op indicatie: Cytoreductieve therapie
Cytoreductieve therapie
Indicaties voor start cytoreductieve therapie en behandeldoel:
- Leeftijd >60 jaar, streef trombocyten waarde < 400 x109/l
- (doorgemaakt) trombo-embolische complicatie, streef trombocyten waarde < 400 x109/l
- Trombocyten > 1500 x 109/l of VvWD, streef verlagen trombocyten en/of remissie VvWD
- Symptomatische splenomegalie, streef verminderen klachten splenomegalie
- ET gerelateerde symptomen die niet verbeteren na start trombocyten aggregatieremming
Opties:
- 1e lijn
- Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Gepegyleerd interferon α 2a (Pegasys®), start dosering 45-90 microgram/week s.c.
- 2e lijn:
- Hydroxycarbamide, start dosering 1dd 500-1000mg oraal
- Gepegyleerd interferon α 2a (Pegasys®), start dosering 90 microgram/week s.c.
- Anagrelide, start dosering 2dd 0,5mg, ieder week te verhogen met 0,5 mg/dag extra op geleide van trombocyten aantal. Maximale dosis 10 mg/dag en 2 mg/gift.
- Combinatie van behandelingen
- Busulfan, start dosering 2-4 mg/dag. (CAVE langdurige cytopenie: controle bloedbeeld en cave leukemogeniciteit)
- Melfalan, start dosering 2 mg 3 x/week. (CAVE cytopenie: controle bloedbeeld en cave leukemogeniciteit)
- Fosfor-32 (32P), dosering 3mCI i.v, zonodig na 3 maanden herhalen. (CAVE leukemogeniciteit)
Trombaferese
Indicatie: Indien in zeer korte tijd het trombocytengetal verlaagd moet worden bij zeer hoge getallen, gecompliceerd door bloedingen of trombotische complicaties.
Primaire myelofibrose
Diagnostische criteria
De herziening van de diagnostische WHO criteria in 2016, laat een indeling zien in een vroeg, pre-fibrotische en een overt stadium.
Diagnostische WHO-criteria (2016)
Diagnostische criteria pre-PMF WHO 2016
| Major criteria: |
|
| Minor criteria: (laboratorium afwijkingen bij 2 opeenvolgende metingen bepaald) |
|
|
Diagnose pre-PMF: 3 major criteria in combinatie met minimaal 1 minor criterium |
| Voetnoten: * : In de afwezigheid van de drie klonale markers, kan het bepalen van andere somatische mutaties behulpzaam zijn bij het aantonen van klonaliteit (bij voorbeeld: ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc) **: BM-fibrose secundair aan infectie, auto-immuun aandoening, chronisch inflammatoire aandoening, hairy cell leukemie of ander lymfatische neoplasie, gemetastaseerde ziekte, of toxische beenmerg afwijkingen. |
Diagnostische criteria overte PMF WHO 2016
| Major criteria: |
|
| Minor criteria: (laboratorium afwijkingen bij 2 opeenvolgende metingen bepaald) |
|
| Diagnostische criteria overte PMF WHO 2016: 3 major criteria in combinatie met minimaal 1 minor criterium |
| Voetnoten: *: In de afwezigheid van de drie klonale markers, kan het bepalen van andere somatische mutaties behulpzaam zijn bij het aantonen van klonaliteit (bij voorbeeld: ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc) **: BM-fibrose secundair aan infectie, auto-immuun aandoening, chronisch inflammatoire aandoening, hairy cell leukemie of ander lymfatische neoplasie, gemetastaseerde ziekte, of toxische beenmerg afwijkingen |
Onderzoek
- Anamnese met aandacht voor algemene klachten, constitutionele symptomen en klachten van splenomegalie (bij voorkeur ook objectiveren met Myeloproliferative Neoplasm-Symptom Assessment Form (MPN-SAF vragenlijsten), bloedingsneiging, cardiovasculaire risico factoren en jicht.
- Lichamelijk onderzoek: bloeddruk, palpatie lever en miltgrootte
- Bloedbeeld (inclusief leukocyten differentiatie), reticulocyten, kreatinine, leverenzymen, LDH, urinezuur, glucose en cholesterol/triglyceriden
- Mutatie bepaling: JAK2V617F, BCR-ABL1 genfusie (ter uitsluiting CML) en bij negatieve testuitslag CALR en MPL mutatie (eventueel bepalingen tegelijkertijd, afhankelijk van werkwijze moleculair laboratorium)
- Bloed-en beenmerg morfologie (bijvoorbeeld leuco-erytroblastair bloedbeeld, tear drop cellen)
- Beenmergbiopt
- Cytogenetica (prognostische betekenis)
- Op indicatie:
- Aanvullende moleculaire diagnostiek (ASXL1, EZH2, TET2, IDH1/IDH2, SRSF2, SF3B1, etc) bij patiënt met mogelijke indicatie allogene stamceltransplantatie (prognostische betekenis, MIPSS70/MIPSS70plus)
- Echo milt bij twijfel over splenomegalie
Behandeling
Algemeen
- Indien asymptomatisch geen (cytoreductieve) therapie
- Thrombocytenaggregatie remming tenzij trombopenie (trombocyten aantal <50 x109/l). Overwogen kan worden te kiezen voor een avond dosering i.p.v. een ochtend dosering
- Overweeg allo-SCT bij patiënten <70 jaar met hoog risicoprofiel volgens MIPPS70 of MIPPS70plus V2.0 (zie onder risico groepen)
- Voorafgaand aan allo-SCT, of indien er geen indicatie of mogelijkheid is tot het doorleiden naar allo-SCT overweeg cytoreductieve therapie:
- Indicaties voor start cytoreductieve therapie en behandeldoel:
- (Doorgemaakt) trombo-embolische complicatie, streef trombocyten waarde <400 x109/l
- Verworven van Willebrand ziekte (VvWD), streef verlagen trombocyten en/of remissie VvWD
- Symptomatische splenomegalie
- PMF gerelateerde symptomen
- Overweeg cytoreductie:
- Progressieve myeloproliferatie (leucocytose >25 x109/l), streef naar leucocyten <15 x109/l
- Leeftijd >60 jaar, streef trombocyten waarde <400 x109/l
Risicogroepen
De DIPSS-plus score is voor alle PMF-patiënten bruikbaar voor het inschatten van de levensverwachting (overall survival).
De MIPSS70 en de MIPPS70plus V2.0 score zijn bedoeld voor het inschatten van de levensverwachting ter identificatie van kandidaten voor allogene stamcel transplantatie (70 jaar of jonger).
Dynamic International Prognostic Scoring System Plus (DIPSS Plus)
| Parameter | Score |
| Leeftijd > 65 jaar | 1 |
| Constitutionele symptomen | 1 |
| Hb < 6,2 mmol/L | 1 |
| Leukocytose > 25 x 109/L | 1 |
| Blasten perifeer bloed ≥ 1% | 1 |
| Trombocytopenie (< 100 x109/L) | 1 |
| Cytogenetica: complex, +8, -7/7q-, i(17q), 5/5q-, 12p-, inv(3), of 11q23 rearrangement | 1 |
| Erytrocyten transfusie afhankelijkheid | 1 |
| Risico indeling DIPSS Plus | Risicofactoren | Mediane overall survival (maanden) |
| laag | 0 | 185 |
| intermediair-1 | 1 | 78 |
| intermediair-2 | 2-3 | 35 |
| hoog | ≥ 4 | 16 |
MIPSS70, prognostische score met incorporatie van de moleculaire diagnostiek
| Parameter | Score |
| Constitutionele symptomen | 1 |
| Hb <6,2 mmol/L | 1 |
| Leukocytose >25 x 109/L | 2 |
| Blasten perifeer bloed ≥ 2% | 1 |
| Trombocytopenie (<100 x109/L) | 2 |
| Beenmerg fibrose graad 2 of graad 3 | 1 |
| Hoog-moleculair risico: 1 HMR afwijking* | 1 |
| 2 of meer HMR-mutaties* | 2 |
| Afwezigheid CALR-type 1 mutatie | 1 |
| * ASXL1, SRSF2, IDH1/2, EZH2 | |
| Risico indeling MIPSS70 | Score | Mediane overall survival (jaren) |
| laag | 0-1 | 27,7 |
| intermediair | 2-4 | 7,1 |
| hoog | > 5 | 2,3 |
MIPSS70plus v2.0, prognostische score met incorporatie van moleculaire diagnostiek en cytogenetica
| Parameter | |
| Constitutionele symptomen | |
| Anemie – milde anemie * – ernstige anemie ** |
* man: Hb >5,6 en <6,8 mmol/L; vrouw Hb >5,0 en <6,2 mmol ** man: Hb <5,6 mmol/L; vrouw Hb < 5,0 mmol/L |
| Blasten perifeer bloed ≥ 2% | |
| Mutaties – hoog-moleculair risico: 1 HMR afwijking *** – 2 of meer HMR-mutaties *** – afwezigheid CALR-type 1 mutatie |
*** ASXL1, SRSF2, IDG1/2, EZH2 |
| Karyotype – unfavourable karyotype **** – very high risk karyotype ***** |
**** Ieder afwijkend karyotype, behalve: 20q-, 13q-, +9 chromosoom 1 translocatie/duplicatie, -Y of geslachtschromosoom afwijking anders dan -Y ***** Very high risk (VHR): enkel of meervoudige afwijkingen van -7, i(17q), inv(3)/3q21, 12p-/12p11.2, 11q-/11q23, of andere autosomale trisomieën maar geen +8/+9 (bijv: +21, +19) |
| Risico indeling MIPSS70-plus v2.0 | score | mediane overall survival (jaren) |
| zeer laag | 0 | >10 (niet bereikt in studie) |
| laag | 1-2 | 10,3 |
| intermediair | 3-4 | 7 |
| hoog | 5-8 | 3,5 |
| zeer hoog | >9 | 1,8 |
Myelofibrosis Transplant Scoring System (MTSS)
Voor een inschatting van het transplantatierisico en uitkomst kan gebruik gemaakt worden van de Myelofibrosis Transplant Scoring system (MTSS score)
| Parameter | score |
| Leeftijd > 57 jaar | 1 |
| Karnofsky score < 90% | 1 |
| Non-CALR/MPL driver mutatie | 2 |
| ASXL1 mutatie | 1 |
| Leukocyten aantal >25 x109/L voor allogene stamceltransplantatie | 1 |
| Trombocyten aantal <150 x109/L voor allogene stamceltransplantatie | 1 |
| HLA-mismatch unrelated donor | 2 |
| Risico indeling | score | 5-jaars NRM (%) | Survival % (5-jaar) |
| Low | 0-2 | 10 | 83 |
| Intermediate | 3-4 | 22 | 64 |
| High | 5 | 36 | 37 |
| Very high | 6-9 | 57 | 22 |
| NRM: non-relapse mortality | |||
Behandeling
De behandeling van PMF is met name symptomatisch. Afhankelijk van de risicoscore en leeftijd van de patiënt kan een allogene stamceltransplantatie overwogen worden (zie hoofdstuk transplantatie indicaties; indicatie bij intermediair of hoog risico volgens MIPSS70 en/of MIPSS70plus v2.0)
Indien behandelindicatie
1e lijn:
- Indien symptomatische splenomegalie en/of trombocytose/leukocytose: Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Indien alleen trombocytose of leukocytose: Gepegyleerd interferon α 2a (Pegasys®), start dosering 45-90 microgram/week s.c.
- Indien PMF gerelateerde symptomen en/of symptomatische splenomegalie: Ruxolitinib. Start dosering 2dd 20 mg, afhankelijk van trombocytenwaarde startdosering aanpassen. Aandachtspunten bij start ruxolitinib zijn weergegeven in tabel 1)
2e lijn en verder:
- Indien symptomatische splenomegalie en/of PMF gerelateerde symptomen: Ruxolitinib. Start dosering 2dd 20 mg, afhankelijk van trombocytenwaarde startdosering aanpassen. Aandachtspunten bij start ruxolitinib zijn weergegeven in tabel 1
- Indien symptomatische splenomegalie en/of PMF gerelateerde symptomen EN Ruxolitinib intolerantie of refractariteit: fedratinib 1 dd 400 mg. Aandachtspunten bij start ruxolitinib zijn weergegeven in tabel 2. De definities ruxolitinib intolerantie en refractairiteit staan in tabel 3 weergegeven.
- Hydroxycarbamide, start dosering 1dd 500-1000 mg oraal
- Gepegyleerd interferon α 2a (Pegasys®), start dosering 45-90 microgram/week s.c.
- Combinatie van behandelingen
- Indien alleen trombocytose: Anagrelide, start dosering 2dd 0,5mg, ieder week te verhogen met 0,5 mg/dag extra op geleide van trombocyten aantal. Maximale dosis: 10 mg/dag en 2,5 mg/gift.
- Indien met name anemie en splenomegalie: Lenalidomide, monotherapie of in combinatie met corticosteroïden (bijvoorbeeld lenalidomide 1 dd 10-15 mg + prednison 3 maanden 1 dd 20 mg, daarna 1 dd 10 mg).
|
|
|
|
|
|
|
|
|
| Refractairiteit | Ruxolitinib therapie > 3 mnd met <10% miltvolume reductie of <30% milt grootte t.o.v. baseline |
| Recidief/progressie ziekte | Ruxolitinib therapie > 3 mnd met verlies van milt respons |
| Intolerantie | Ruxolitinib therapie > 28 dagen met ontstaan van erythocyten transfusie-afhankelijkheid (> 2 E/maand gedurende 2 mnd), of > gr 3 trombopenie en/of anemie en/of bloeding en/of hematoom |
Overige opties:
- In geval van anemie:
- Danazol, 200-600 mg per dag p.o., maximaal 3 maanden. Indien hoge dosering (> 400mg, dan verdelen over 2 giften)
- Erytropoëtine: Bij een niet-transfusie afhankelijke anemie is de kans op respons 90%, bij een transfusie afhankelijke anemie 40%. Bij een laag serum erytropoëtine is de kans op succes ~65% bij een normaal serum erytropoëtine ~15%. Advies: Overweeg epo toediening indien anemie en lage endogene epo. Cave: toename splenomegalie
- Erytrocyten transfusies
- T.a.v. splenomegalie met ernstige mechanische bezwaren: cytoreductie of JAK2 remmers.
- Hydroxyurea, 2 x 500-1000 mg/dag p.o., op geleide bloedbeeld.
N.B.: Frequente controle noodzakelijk i.v.m. ernstige cytopenieën. - JAK2 remmers: ruxolitinib/fedratinib (zie aldaar).
- Indien refractaire ziekte of onacceptabele bijwerkingen ander cytoreductie en beperkte levensduur: overweeg busulfan (1 dd 2 mg, cave pancytopenie), of eventueel melfalan (3x per week 2 mg)
- Hydroxyurea, 2 x 500-1000 mg/dag p.o., op geleide bloedbeeld.
Splenectomie kan overwogen worden indien cytoreductieve therapie niet succesvol en:
- progressieve transfusiebehoefte;
- ernstige hemolyse (geen reactie op steroïden);
- ernstige trombocytopenie;
- ernstige mechanische bezwaren;
- miltinfarct (recidiverend);
Bij overgang naar AML: zie acute leukemie;
Bij leeftijd ≤ 70 jr: overweeg allo-SCT (RIC).
Indien splenectomie gecontra-indiceerd
- Overweeg miltbestraling (mediane respons duur ongeveer 6 maanden)
MPN en splanchnicus trombose
- cytoreductieve therapie, streef Ht <0,45 l/l, trombocyten aantal <400 x 109/l en overweeg (gezien de relatie tussen leukocytose en trombose) streef leukocyten <15 x 109/l
- levenslang therapeutische antistolling (tenzij contra-indicatie)
- start in de stabiele fase trombocyten aggregatieremming naast therapeutische antistolling indien laag bloedingsrisico (cave oesophagus varices)
Zwangerschap en myeloproliferatieve syndromen
- Behandeling bij geen risicofactoren (zie tabel 4):
- Acetylsalicylzuur 80 mg/Carbasalaatcalcium 100 mg/dag
- Profylactische dosering laagmoleculair gewichtsheparine (LMWH) in het kraambed
- Streef naar Ht < 0.40 l/l indien PV (middels flebotomie en/of gepegyleerde interferon-α)
- Trombocyten < 1000 x109/l (middels gepegyleerde interferon-α)
- Behandeling bij risicofactor (zie tabel 4):
- Acetylsalicylzuur 80 mg/Carbasalaatcalcium 100 mg/ dag
- Trombocyten < 400 x109/l (middels gepegyleerde interferon-α)
- Streef naar Ht < 0.40 l/l indien PV (middels flebotomie en/of gepegyleerde interferon-α)
- LMWH gedurende gehele zwangerschap en het kraambed. Therapeutische dosering LMWH indien behandeling met orale antistollingstherapie voor de zwangerschap, anders profylactische dosering LMWH.
- Acetylsalicylzuur 80 mg/Carbasalaatcalcium 100 mg/ dag
| Risico factor: |
| Trombose in voorgeschiedenis |
| Bloeding in voorgeschiedenis |
| Bij eerdere zwangerschap ≥3 spontane abortus binnen eerste trimester |
| Bij eerdere zwangerschap spontane abortus in tweede of derde trimester |
| Bij eerdere zwangerschap intra uteriene sterfte |
| Bij eerdere zwangerschap pre eclampsie <37 weken, intra-uterine groei achterstand of aanwijzingen voor placenta dysfunctie |
| Bij eerdere zwangerschap bloeding of post-partum bloeding met transfusie noodzaak |
| Bij huidige zwangerschap abnormale flow in de uteriene arteriën op 20/24 weken |